Joseph J. Eron MD Professor of Medicine University of North Carolina at Chapel Hill
The CROI meeting in Seattle this year was again a terrific mix of basic, translational, clinical and epidemiological research. This year perhaps we are seeing more clinical data than we have seen in the last few CROI with some terrific late breaker presentations.
Antiretroviral treatment – where we are now
There were two very nice poster presentations from Thibaut Davy-Mendez who is a PhD candidate at UNC. Working with data from our UNC CFAR HIV Clinical Cohort he described trends in antiretroviral treatment and HIV resistance, prevalence and incidence over the last several years.
In his presentation on Increased Persistence Of Initial Art With INSTI-containing Regimens [Davy et al Abstract 465] he demonstrates the dramatic shift to initial therapy with integrase- inhibitor based regimens in the last several years. By 2014 over 80% of new treatment starts were with INSTI-based regimens in our cohort. This shift has resulted in substantial differences in persistence of first-line therapy (by that we mean how long someone stays on their first regimen before switching or discontinuing their anchor agent) and also in the rates of virologic failure. Davy-Mendez showed that the median time to moving away from or stopping INSTI-based therapy was greater than 6 years and that patients on an INSTI-based therapy were significantly and substantially less likely to stop or switch from INSTI-based therapy compared to NNRTI based therapy (HR = 0.49 (0.35, 0.69). Those patients started on INSTI-therapy were also less likely to experience virologic failure. These data are only likely to improve as much of the data were captured before single tablet integrase inhibitor therapy became the norm.
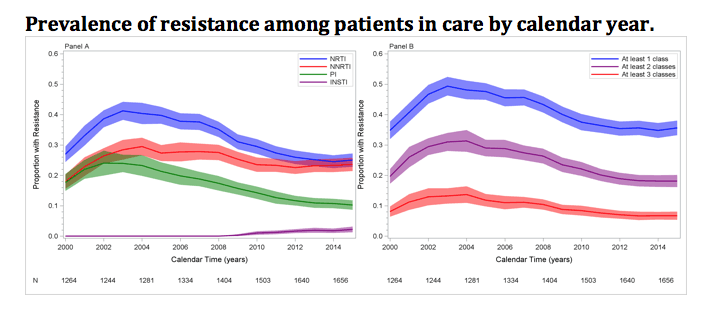
Davy-Mendez also looked at prevalence of resistance in our cohort in Low Prevalence Of HIV Drug Resistance With Modern Agents [Davy et al Abstract 483]. In this analysis he did two things: first he looked at prevalence of resistance in all patients who were active in the cohort in a given year. All previous and current resistance tests were used and resistance was counted even if the patient’s viral load was suppressed. For example if in 2006 a patient has viremia and a resistance test that show NRTI, NNRTI and PI resistance that information is captured for that year. If in 2007 she suppressed on raltegravir, etravirine and darunavir/r (like many patients did) that resistance burden still exists in the cohort even though she is suppressed on therapy. The resistance remains in every year she stays in the cohort. The second analysis looks only at resistance in patients who have viremia in a given year and the resistance assessment includes all past tests (cumulative resistance). So if our patient listed above rebounds in 2010 and has INSTI-resistance documented her cumulative resistance would include 4 classes. You can think of this like an iceberg of resistance. The first analysis is the whole iceberg and gives you an idea of the total resistance burden – hopefully most in underwater (i.e. suppressed). The second analysis is the part of the iceberg that is visible – so circulating resistance in viremic patients. Patients who are lost to follow-up, move away or die no longer contribute to the resistance analysis in the subsequent year. Perhaps a picture is worth 1000 words
This figure is the whole iceberg (notice the y-axis only goes to 60%). We see that the burden of resistance in our cohort (and I suspect most cohorts in the US) is declining. PI and NRTI resistance has declined substantially as has 1-, 2- and 3-class resistance. Fortunately integrase resistance remains very low, less than 5% of patients in the cohort (recall that we are a referral center for much of North Carolina so we may be enriched for “Viking” like patients1).
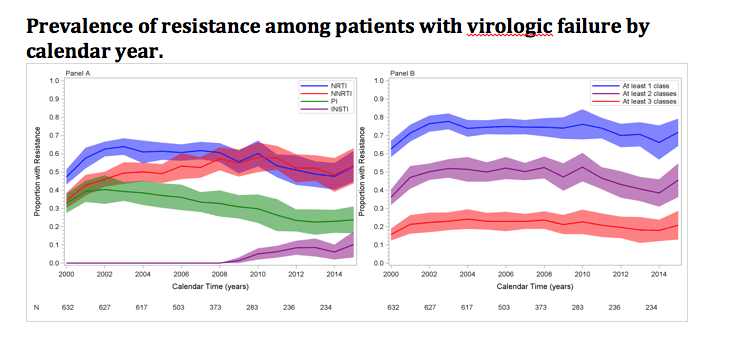
The tip of the iceberg looks like this:
Here the story is a little different – the proportion with resistance is higher and the downward trends are not nearly as obvious (except for maybe protease inhibitor resistance). The good news is that viremic patients are a small proportion of our total patient populations (approximately 10% or less). We also looked at patient who started therapy with modern preferred regimens (2007 to 2014). In this group emergent resistant virus was even much less common – with resistance to at least one drug in 2 classes emerging in only 5% of 685 patients and resistance to one or more drugs in 3 classes emerging in only 1%. However resistance is likely to be with us as long as we have HIV and we are treating people who are – like us – human.
PS – Huldrych Gunthardt and the Swiss do these types of analyses really well and I urge you to read the two papers by Scherrer et al cited here2,3.
We need a new drug or a new strategy – or do we?? Some exciting new antiretroviral treatment data were presented at CROI at several levels of development.
Novel combination of InSTI and NNRTI for Maintenance of Suppression
There were two large phase III randomized antiretroviral treatment trials presented.
In the SWORD 1 and 2 studies a novel combination of an INSTI (dolutegravir) and an NNRTI (rilpivirine) were studied in patients suppressed on a first or second regimen with no previous history of resistance or virologic failure [Libre et al Abstract 44LB]. This study is the first successful large comparative study of a two-drug combination maintenance therapy that did not include a boosted protease inhibitor (or a nucleoside analogue). Over 1,000 participants were randomized to either stay on their initial 2NRTI plus anchor agent regimen or switch to dolutegravir 50 mg and rilpivirine 25 mg once daily. Fifty-four percent of participants were on a NNRTI at baseline, approximately 26% were on a PI and 20% were already on an INSTI based regimen. Most were on TDF (approximately 72%). The study was open label and patients with hepatitis B were excluded. Ninety-five percent of participants in each arm were < 50 c/mL at 48 weeks and only 3 participants (<1%) and 6 participant (1%) had virologic non-response in the DTG/RPV and continuation ART arms respectively. There were more discontinuations due to AE in the experimental arm (16 (3%) vs. 2 (<1%)), which is not too surprising given that the median time on stable ART at the time of randomization was > 4 years and that the study was open-label and a new adverse event may have been more likely to be attributed to the experimental therapy than to chronic stable therapy. Supporting this conjecture is that fact that overall AE and serious AE occurred at similar rates in the two arms, more AE’s were attributed as drug related in the DTG/RPV arm. Most were grade 1-2. Only two participants in each arm met protocol defined virologic failure (for some reason termed “confirmed virologic withdrawal”). One participant on the DTG/RPV arm had emergence of a virus with a mixture at K101K/E, which is an NNRTI resistance mutation. However this mutation did not confer phenotypic resistance to RPV, the patient’s rebound viral load was > 1,000,000 (suggesting non-adherence) and the participant re-suppressed on DTG/RPV before therapy could be changed. No integrase resistance mutations were identified.
Implications: Overall an excellent result – high success, minimal virologic non-response and no clear resistance emergence in over 500 patients treated with this simple, low milligram, likely to be co-formulated treatment that will have some but not many drug-drug interactions and no renal clearance. However this was a very conservative study. Participants could have had no virologic failure, no history of transmitted K103N, PI or NRTI mutations despite the fact that the regimen should not be impacted by NRTI or PI resistance and likely not transmitted K103N, which was allowed in the switch studies using rilpivirine/TDF/FTC . Participants with more than one previous treatment switch, even if they remained suppressed, were also excluded. So one might ask, “Who will receive this combination?” Pill size will be important for some patients. Patients on boosted PI who would like simplification would also likely consider this – though many of these patients have already switched therapy. Prior to the introduction of TAF this would have been perhaps an optimal therapy for patients with modest renal dysfunction but if patients are already on a TAF based therapy perhaps only those with more substantial renal compromise (eGFR in 30 mL/min range) would be considered for switching. Many clinicians have patients suppressed on therapy who are on more cumbersome therapy or therapy with potential for longer term toxicity who have NRTI or PI mutations. Such patients might benefit from a switch to DTG/RPV though this was not a group that was studied in SWORD 1 and 2. Whether a such a patient should switch will likely be an individual patient/clinician decision based on the overall risk:benefit ratio of that patient.
We should also note that another simple two-drug combination DTG plus 3TC might also be considered in some of these switch situations. A small non-comparative study of switching stably suppressed patients to DTG/3TC was also presented at CROI. In that study, called Lamidol [Joly et al Abstract 458], patients suppressed on 2 NRTI and a third drug first had their third drug switched to dolutegravir for 8 weeks and if they remained suppressed they entered second phase in which participants had their two NRTI simplified to 300 mg of 3TC. Of 104 participants who entered the second part of the study 101 remained suppressed at 48 weeks with only one virologic failure. This two-drug combination is being studied in two very large (700 participants each) actively enrolling randomized trials, called Gemini 1 and 2, in treatment naÏve patients (clinicaltrials.gov). We won’t have results of these trials for over a year but clinicians may also be thinking about this relatively simple two-drug combination as a switch strategy in the appropriate clinical context.
The first phase III study of the NNRTI doravirine, called DRIVE, was presented in Seattle [Molina J-M et al Abstract 45LB]. Doravirine is a once daily NNRTI that does not have an interaction with proton pump inhibitors, did not appear to have substantial CNS side-effects in a phase II study compared to efavirenz4, has activity against the most common NNRTI-resistance mutations5 and will likely have limited drug-drug and food interactions6. This study, in treatment naÏve participants, compared doravirine to darunavir/ritonavir each patient with either TDF/FTC or ABC/3TC in a randomized double blind fashion. The 766 participants who entered had a choice of NRTI (88% chose TDF/FTC) and had to take 4 pills (doravirine 100 mg with DRV and RTV placebos or doravirine placebo with active DRV and RTV each plus the 2 NRTI tablet). The results at 48 weeks demonstrated that doravirine was non-inferior to DRV/r with 84% vs. 80% < 50 c/mL in the FDA snapshot analysis. CD4 cell count responses were good and similar between the two arms and doravirine responses were consistent in the populations of participants with baseline HIV RNA > 100,000 and > 500,000 c/mL. There were 19 protocol defined virologic failures in the doravirine arm and 24 in the DRV/r arm. In addition, some patients also left the study with HIV RNA > 50 c/mL who had not yet met the definition of virologic failure. Genotype resistance testing was attempted in both these groups. Not surprisingly no participants developed PI or NRTI resistant virus in the DRV/r arm. Remarkably, only a single patient in the doravirine arm (out of 383) developed drug resistance mutations that led to doravirine and FTC/3TC resistance, suggesting a higher barrier to resistance emergence for this NNRTI. Rash and neuropsychiatric adverse events occurred at similar rates in the two arms and overall AE’s were similar with numerically less diarrhea in the doravirine arm (14 vs. 22%). There were relatively few Grade 3 or 4 laboratory abnormalities and not surprisingly the lipid profile of doravirine was more favorable than with DRV/r.
Implications: Cross study comparisons are fraught with potential bias – though it is difficult not to make them. The overall result of suppression at 48 weeks (84 vs. 80%) in the DRIVE study is lower than we have seen with recent integrase inhibitor based studies in which 48 week suppress rates on the integrase inhibitor arms are in the 90% range7-10. One issue with the current study is the regimens were blinded and participants had to take 4 pills a day which may have impacted the retention rate on the study. Approximately 10% of participants in each arm left the study for reasons other than an adverse event and prior to reaching protocol defined virologic failure. Further data are expected as a head-to-head comparison with fixed dose combination efavirenz/TDF/FTC is fully accrued and should give initial results soon and a switch study from efavirenz-based therapy is recruiting (clinicaltrials.gov). Given the results of this first study, doravirine with 2 NRTI is non-inferior to one of the DHHS first-line initial regimens for treatment naÏve patients (DRV/r) at 48 weeks. However, most guidelines and most initial treatment starts in many clinics in the developed world are moving toward or have already embraced integrase inhibitors as the anchor for first line therapy. Where doravirine will fit into that equation is not clear. If data consistent with a favorable CNS profile continue to accrue that could be an advantage of this drug and it will likely be approved as a single agent and will therefore have the flexibility to be partnered with any of the two-drug NRTI combinations. According to Merck a fixed dose formulation with TDF and FTC is in development.
Paul Sax and his colleagues presented Phase II data on the newest integrase inhibitor, bictegravir [Sax P, el al Abstract 41]. Bictegravir is a very promising agent. This INSTI has in vitro and in vivo potency similar to dolutegravir and very favorable pharmacokinetic characteristics that allow once a day dosing with resulting trough levels many fold higher than the protein adjusted IC95. Bictegravir has activity against most INSTI-resistant viruses that are selected for by raltegravir and elvitegravir but, to date, we have seen very little clinical data about this agent despite the fact that four phase III studies are now fully enrolled. In Paul’s presentation we saw the first comparative “longer-term” 48-week data in a randomized comparison to dolutegravir both paired with TAF/FTC in treatment naÏve patients. This is a phase II study so the number of participants in each arm is small and with a 2:1 randomization there were 65 participant randomized to the bictegravir arm and only 33 participants randomized to dolutegravir. Both treatments did very well and after 48 week 97% of participants randomized to the bictegravir arm were < 50 c/mL by the FDA snapshot analysis. Only 2 participants were not successful. One discontinued due to a skin-related adverse event and the other was a virologic failure. There were only 3 virologic failures in the 98 randomized patients – none developed resistance.
Implications: What makes bictegravir potentially special is that it does not require boosting to have excellent PK exposure and it is in development as a co-formulation with TAF and FTC with a total milligram burden of < 300 mg – likely translating into a small pill size. Like dolutegravir drug-drug interactions should be relatively minor. It does look like from Paul Sax’s presentation that bictegravir will increase creatinine slightly presumably through a similar mechanism to dolutegravir and cobicistat. The interaction of bictegravir with metformin appears to be less than with dolutegravir [Zhang H et al Abstract 40].
The proof, of course, will be in the results of the Phase III trials. In treatment naÏve patients there is a comparison similar to the Phase IIB trial – bictegravir/TAF/FTC fixed dose combination (FDC) vs. dolutegravir plus TAF/FTC in a blinded (3 pills each) randomized trial of approximately 645 participants. Also in treatment naÏve participants the second trials compares bictegravir/TAF/FTC vs. Dolutegravir/ABC/3TC both fixed dose combinations, again blinded (2 pills a day) and randomized (approximately 630 participants). Then there are two switch studies for suppressed patients. One enrolling patients on boosted atazanavir or darunavir with either TDF/FTC or ABC/3TC and randomizing participants to stay on the boosted-PI 2 NRTI regimen or switch to bictegravir/TAF/FTC FDC. This is an open-label study with approximately 520 enrolled participants. The second switch studied enrolled patients on dolutegravir with ABC/3TC and randomized them in a blinded fashion to dolutegravir/ABC/3TC FDC or bictegravir/TAF/FTC FDC. Over 560 participants were enrolled. All these studies will reach their primary endpoint sometime in 2017. We look forward to those results. If this fixed dose integrase inhibitor combination is successful then we will have an integrase-based single tablet without boosting, which can be used in hepatitis B and without HLA B5701 testing.
Ibalizumab is a monoclonal antibody that has been in development for many years with the first clinical study published eight years ago11. The antibody blocks HIV replication by binding to the second domain of CD4 molecule preventing HIV entry. Ibalizumab has activity against both R5 and X4 viruses and does not appear to affect CD4 cell function. In its present formulation it has to be administered by IV infusion. At CROI we saw the 24-week results of the very small (40 patient) phase III study [Lewis S et al Abstract 449LB]. The study follows the basic design for antiretrovirals being developed for patients with limited treatment options. At screening patients were required to have resistance to at least one drug in three different antiretroviral classes and have a least one fully active agent to add to an optimized background regimen. Patients with highly resistant virus (35% resistant to multiple agents in 4 or more classes) were enrolled. After entry participants were observed over a 7 day period. They then received an IV infusion of ibalizumab as functional mono-therapy and the results at 7 days is actually the primary endpoint of the study. Treatment is then optimized at day 7 and in this study some patient were able to use a second novel agent fostemsavir, which is an oral attachment inhibitor. Ibalizumab infusion continues 2 weeks along with the optimized background and the 24 week results were presented. Forty patients were enrolled with mean CD4 cell count of 150 cells/mm3 at baseline. 83% had > 0.5 log10 decline and 60% had a > 1.0 log10 decline over 7 days. The longer term, 24 week results, reflected the extensive pre-entry resistance with only 43% having an HIV RNA < 50 c/mL at week 24 and in participants with < 50 CD4 cells/mm3 at baseline less than 20% were < 50 c/mL at 24 weeks. Participants who received fostemsavir actually had numerically poorer outcomes than those who had an active agent available in an existing class though no formal statistical analysis was undertaken. Ibalizumab appeared safe. Eight of the 9 participants who discontinued had < 50 CD4 cells/mm3 at baseline (4 were deaths).
Implications: There are patients in our clinics who will likely benefit from ibalizumab. They mirror the patients enrolled in this trial with highly antiretroviral drug resistance virus and limited treatment options. Fortunately these patients are uncommon and are typically patients who have been in care for prolonged periods and received sequential mono-therapy over the last 3 decades as we learned how to use antiretrovirals effectively and new agents appeared one at a time. At present the administration requires IV infusion and if approved by the FDA will likely be labeled as an every 2 week infusion given the phase III study design. The antibody has relatively modest potency with average short-term log reduction in the 1.0 log10 range. Intramuscular injections every 2 weeks and every 4 weeks are being explored [Lin HH el al Abstract 438] and appear to give similar exposure. According to the Theratechnologies website an FDA NDA filing is planned.
Clinical data on two other monoclonal antibodies that block HIV-1 entry into cells were also presented at CROI. One, PRO-140 has been around in development for a very long time. This antibody binds to CCR-5 and block entry of R5 viruses (like the small molecule maraviroc). The study presented was a long-term follow-up of a subset of patients, with R5 virus, who were on successfully suppressive oral therapy and had switched to single-agent therapy with this antibody delivered as a weekly subcutaneous injection [Lalezari et al Abstract 437]. The original study started with 31 patients who were treated for 12 weeks and 16 of these participants were entered into long-term follow-up. Over two years, 10 of the sixteen maintained suppression on this single self-administered SQ agent. One participant has been suppressed for 41 weeks. The other 9 have been suppressed for > 105 weeks at the time of the presentation. We were told that the 6 patients who did not stay suppressed did not rebound with X4 or dual tropic virus and that the reason for rebound was likely drug exposure but the primary data were not presented on the poster. There were very few adverse events that were attributed to the medication with a single unrelated serious adverse event and no severe adverse events in the 16 participants.
Implications: Obviously these are very preliminary data in a select population (remember that it is really 10 of 31 who started the mono-therapy > 2 years earlier) and there is no control group. On the other hand two years on a single drug (even if given subcutaneously) is an eye-opening result and I spoke with one of the investigators who said his patient tolerated the therapy very well and are happy with self-administering the antibody. One could imagine that patients who are on complex oral therapies, that don’t include maraviroc and who have R5 virus using an archive DNA assay could try this approach that would be completely orthogonal [completely different – not overlapping – so in this case no chance of selecting for cross resistance] to their oral regimen. The risk, of course, would be viral rebound but presumably restarting oral therapy would be successful.
A study to test this hypothesis is ongoing. In PRO140_CD03, 300 participants with HIV RNA suppressed on oral antiretroviral therapy who have CCR5-tropic HIV-1 infection are being enrolled and will receive PRO 140 as long-acting, single-agent maintenance therapy for 48 weeks. On clinicaltrials.gov that study is listed as enrolling.
There is also a standard phase III study (like the ibalizumab study described above) for patients with highly drug resistant virus that is also enrolling (Pivotal Combination Study: PRO140_CD02).
One other monoclonal antibody that is being studied in HIV-1 infected people was presented. This antibody, UB-421, like ibalizumab binds to the CD4 molecule blocking HIV entry although it binds to the first domain of CD4 not the second domain which may have implications for CD4 cell function. The authors presented a study, similar to the PRO-140 study in which patient who were suppressed on oral therapy were switched to intravenous infusion of UB-421 either weekly (14 participants) or every other week (15 participants) for 8 infusions [Wang CY, et al. Abstract 450BLB]. Remarkably all the participants remained suppressed for the 8- and 16-week periods respectively and then stayed suppressed or re-suppressed (several chose not to restart oral therapy right away) when oral therapy was restarted. This monoclonal antibody is at a much earlier stage of development than either ibalizumab or PRO-140 and there are no open studies listed on clinicaltrials.gov. In the study presented there was an effect on the level of T regulatory cells in the blood of the participants suggesting there may be some effect on T-cell trafficking which will be important to better understand if this antibody moves forward in development.
Capsid Inhibitor
Winston Tse from Gilead presented data on perhaps one of the most novel classes of antiretrovirals to come along in a while, capsid inhibitors [Tse W, et al Abstract 38]. The capsid is what coats the HIV genetic material and sits under the viral envelope. It is essential in several steps in the HIV life cycle including viral assembly, uncoating after entry (which can’t be done haphazardly or the virus goes down the proteasome drain) and transport to the nucleus of the pre-integration complex. Targeting the capsid is tough because it is not an enzyme that has a specific binding site but could be rewarding given that multiple steps in the life cycle may be blocked. The prototype capsid inhibitor presented is extremely potent (pico-molar range – 10-12 concentration) and targets a very conserved region so that resistance variants may be hard to select for (though NOT impossible) or may have diminished fitness. This class of capsid inhibitors may have a very long half-life in people. In an animal model (rat) a single sub-cutaneous injection sustained levels above paIC95 for 10 weeks. These are early days however and no data are available yet for in vivo models of HIV/SIV infection (i.e. monkeys or humanized mice) and no human PK data were presented. We are hearing more about very long acting agents and, at least for treatment, we are likely to need two or more with similar half-lives to combine to create an effective therapy. Capsid inhibitors may be a step in that direction. However, this agent is in early pre-clinical development and has not been given to humans yet even in single dose PK studies. An oral form may be difficult to develop.
Finally we did see first presentations of early pre-clinical data on two drugs in existing classes. One was a NRTI (GS-9131)[White K, et al Abstract 436]. You might ask, “Why would we need another NRTI?” This NRTI had impressive in vitro activity against very resistant nucleoside/tide resistant variants including variants with resistance to tenofovir and variants with multiple thymidine analogue mutations (TAMS). Only the uncommon 151 mutation complex led to an increase fold-change in vitro. The presenter, Kirsten White, stated that this drug is scheduled to begin PK studies in people and one could imagine combining this NRTI with other agents, such as a second generation integrase inhibitor, to be used in patients with drug-resistant virus. There was also a presentation on a new protease inhibitor [Link J, et al Abstract 433]. This agent was presented predominantly because in vitro suggested a high barrier to resistance and animal PK data and human liver microsome data suggested limited metabolism and a relatively long half-life that would likely mean that this PI would NOT require boosting with ritonavir or cobicistat and therefore might have the potential for co-formulation. Early days – no human studies have started yet.
Davy T, Napravnik S, Zakharova O et al. Increased Persistence Of Initial Art With INSTI-containing Regimens. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 465
Davy T, Brunet L, Napravnik S, Zakharova O et al. Prevalence Of HIV Drug Resistance With Modern Agents. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 483
Libre J, et al. SWORD 1 & 2: Switch to DTG + RPV Maintains Virologic Suppression Through 48 Weeks, a Phase III Study. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 44LB
Joly V, Burdet C, Landman R, et al, PROMISING RESULTS OF DOLUTEGRAVIR + LAMIVUDINE MAINTENANCE IN ANRS 167 LAMIDOL TRIAL. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 458
Molina J-M, Squires K, Sax P, et al. DORAVIRINE IS NON-INFERIOR TO DARUNAVIR/R IN PHASE 3 TREATMENT-NAÏVE TRIAL AT WEEK 48. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 45LB
Sax P, DeJesus E, Crofoot G, et al. RANDOMIZED TRIAL OF BICTEGRAVIR OR DOLUTEGRAVIR WITH FTC/TAF FOR INITIAL HIV THERAPY. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 41
Zhang H, Custodio J, et al. CLINICAL PHARMACOLOGY OF THE HIV INTEGRASE STRAND TRANSFER INHIBITOR BICTEGRAVIR. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 40
Lewis S, Fressel J, Emu B, et al. LONG-ACTING IBALIZUMAB IN PATIENTS WITH MULTI-DRUG RESISTANT HIV-1: A 24-WEEK STUDY. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 449LB
Lin HH, Lee S, Wang NC, et al. INTRAMUSCULAR IBALIZUMAB: PHARMACOKINETICS, SAFETY, AND EFFICACY VS IV ADMINISTRATION. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 438
Lalezari J, Dhody K, et al. PRO 140 Single-Agent Maintenance Therapy for HIV-1 Infection: A 2-Year Update. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 437
Wang CY, Wong WW, Tsai HC, et al. A Phase 2 Open-Label Trial of Antibody UB-421 Monotherapy as a Substitute for HAART. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 450LB
Tse W, Link J, Mulato A, et al. Discovery of Novel Potent HIV Capsid Inhibitors with Long-Acting Potential. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 38
White K, Margot N, Stray K, et al. GS-9131 is a Novel NRTI with Activity Against NRTI-Resistant HIV-1. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 436
Link J, Cato D, Moore M, et al. Novel HIV PI with High Resistance Barrier and Potential for Unboosted QD Oral Dosing. Conference on Retroviruses and Opportunistic Infections (CROI), February 13-16, 2017, Seattle. Abstract 433
Other References:
1. Eron JJ, Clotet B, Durant J, et al. Safety and Efficacy of Dolutegravir (DTG) in HIV-1 Treatment-Experienced Subjects With Raltegravir-Resistant Virus: 24-Week Results of the VIKING Study. J Infect Dis. Dec 7 2012.
2. Scherrer AU, von Wyl V, Yang WL, et al. Emergence of Acquired HIV-1 Drug Resistance Almost Stopped in Switzerland: A 15-Year Prospective Cohort Analysis. Clin Infect Dis. May 15 2016;62(10):1310-1317.
3. Scherrer AU, Yang WL, Kouyos RD, et al. Successful Prevention of Transmission of Integrase Resistance in the Swiss HIV Cohort Study. J Infect Dis. Aug 01 2016;214(3):399-402.
4. Gatell JM, Morales-Ramirez JO, Hagins DP, et al. Forty-eight-week efficacy and safety and early CNS tolerability of doravirine (MK-1439), a novel NNRTI, with TDF/FTC in ART-naive HIV-positive patients. J Int AIDS Soc. 2014;17(4 Suppl 3):19532.
5. Feng M, Sachs NA, Xu M, et al. Doravirine Suppresses Common Nonnucleoside Reverse Transcriptase Inhibitor-Associated Mutants at Clinically Relevant Concentrations. Antimicrob Agents Chemother. Apr 2016;60(4):2241-2247.
6. Anderson MS, Gilmartin J, Cilissen C, et al. Safety, tolerability and pharmacokinetics of doravirine, a novel HIV non-nucleoside reverse transcriptase inhibitor, after single and multiple doses in healthy subjects. Antivir Ther. 2015;20(4):397-405.
7. Clotet B, Feinberg J, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 48 week results from the randomised open-label phase 3b study. Lancet. Mar 31 2014.
8. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. Nov 7 2013;369(19):1807-1818.
9. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. Jun 27 2015;385(9987):2606-2615.
10. Sax PE, DeJesus E, Mills A, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. Jun 30 2012;379(9835):2439-2448.
11. Jacobson JM, Kuritzkes DR, Godofsky E, et al. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother. Feb 2009;53(2):450-457.